Long‐read sequencing has become a valuable complement to short‐read approaches in metagenomics because single-molecule platforms routinely deliver reads tens of kilobases in length, thereby reducing assembly fragmentation in complex communities. Pacific Biosciences HiFi chemistry illustrates the current balance between length and fidelity, generating reads of ≈25 kb with reported per-base accuracies approaching 99.9 %, which can lessen the need for subsequent polishing. Oxford Nanopore protocols have further extended read-length distributions, with N50 values above 100 kb and maximum reads close to 0.9 Mb, a scale that is able to span large repeat regions and structural rearrangements. Integrating such long reads into metagenomic assemblies has been shown to facilitate recovery of near-complete, single-contig MAGs and to resolve rRNA operons or mobile-element contexts that often remain fragmented in short-read data.
Assembly-free pipelines interrogate host-depleted shotgun reads directly against curated references to deliver rapid taxonomic profiles and gene-centric summaries, offering immediate insight when turnaround time or compute is constrained. Kraken 2 exemplifies this strategy: by adopting minimizer-based, compact hash indexing, it reduces memory usage by ~85% and increases classification speed by roughly five-fold relative to Kraken 1, while preserving high accuracy. In parallel, Abricate provides fast gene screening on FASTA sequences, aligning queries to well-maintained antimicrobial-resistance and virulence catalogs—including the AMRFinderPlus-derived "ncbi" set and VFDB—and reporting per-hit identity and coverage for straightforward presence/absence tallies that complement Kraken 2’s taxonomy.
Assembly-based, genome-centric analysis reconstructs population genomes directly from metagenomic sequencing by (i) assembling reads into contigs (using long-read–aware or hybrid strategies to capture operons, mobile elements and other genomic context), (ii) clustering contigs into metagenome-assembled genomes (MAGs) with modern binning approaches, (iii) evaluating draft genomes with machine-learning quality models, and (iv) placing them into standardized taxonomies and annotating their functions. This genome-resolved workflow delivers culture-independent access to microbial population genomes and their metabolic potential, enabling synteny-aware interpretation, strain-level comparison and ecological inference across environments. Representative implementations include long-read and hybrid assemblers such as metaFlye and hybridSPAdes, self-/semi-supervised binners such as SemiBin2, quality assessment with CheckM2, phylogenomic classification with GTDB-Tk, and orthology-based functional annotation with eggNOG-mapper; together these advances have powered large catalogues of previously uncultured lineages and expanded reference diversity for downstream analyses.
This module turns contigs into an interpretable, study-wide catalogue of genes in a few clear steps: (1) predict ORFs on assembled contigs with metagenome-aware callers (e.g., MetaProdigal/Prodigal) to capture coding sequences and translation starts; (2) build a non-redundant protein set by clustering translated sequences at user-defined identity thresholds with scalable tools such as CD-HIT or MMseqs2; (3) assign functions at scale using eggNOG-mapper v2, which couples orthology-based annotation with efficient domain discovery and optional GFF decoration for downstream summaries (GO/COG, domains); and (4) deepen enzyme families of interest with focused resources like dbCAN3, which augments CAZyme calls with subfamily HMMs and gene-cluster/PUL context to suggest likely glycan substrates. Together, these steps produce concise per-gene and per-pathway tables that support ecological interpretation, metabolic reconstruction, and cross-sample comparison.

Find data fast from the home page—search by sample keywords, accession IDs, or genome (MAG) names.
Search result page
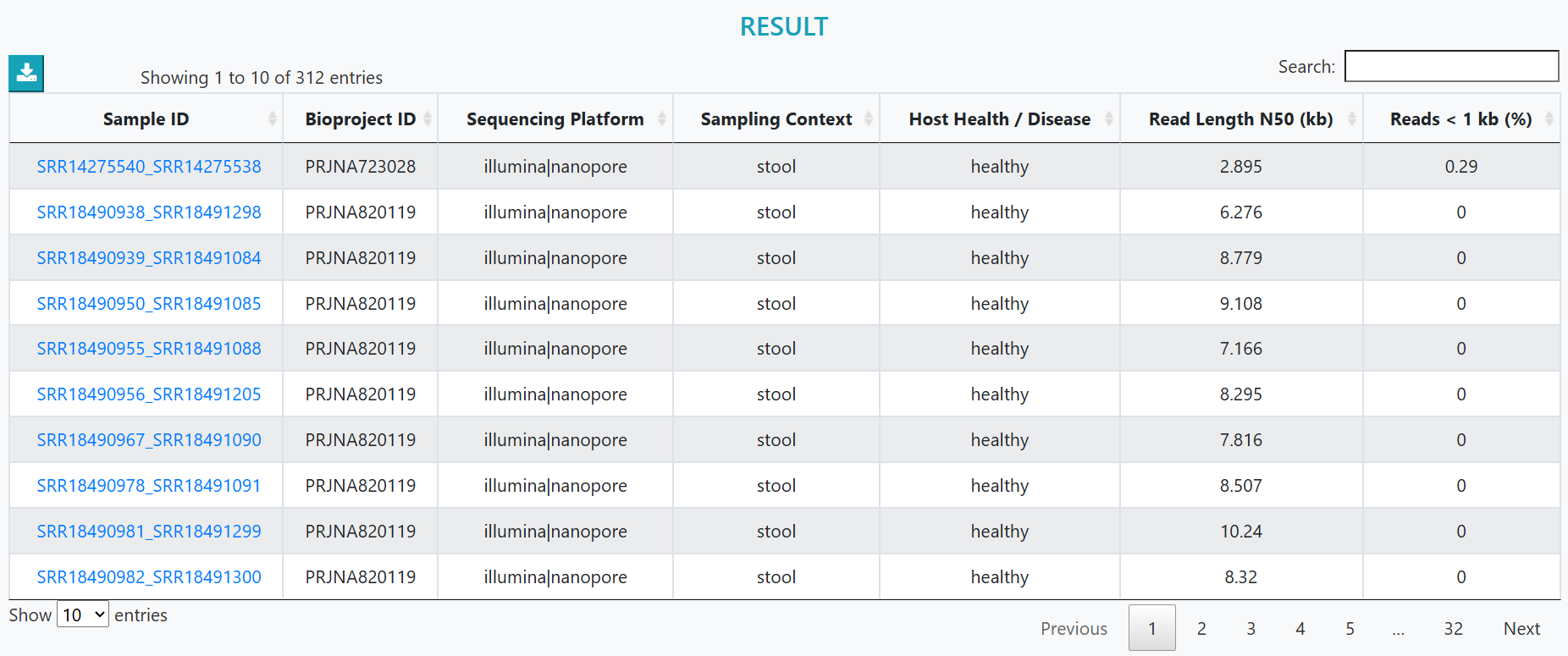
Results that match your filters appear below. Select a Sample ID to open its details.
Detailed information page
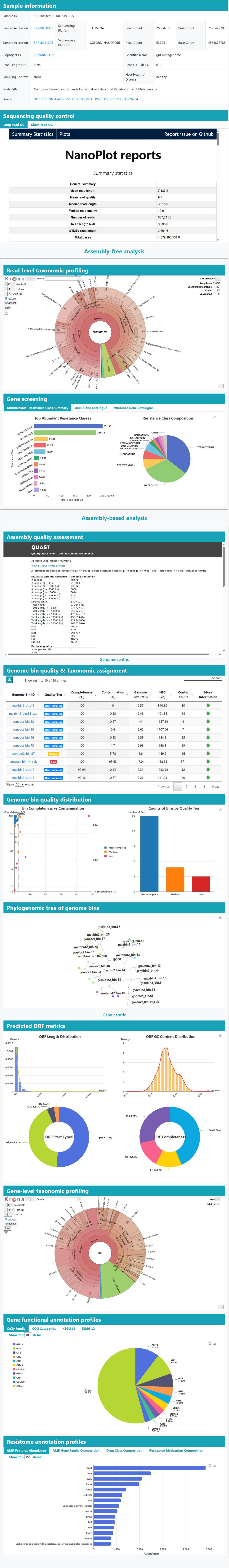
>Detailed information for metagenomic samples
(A) Sample at a glance: Key facts first (Sample ID, run accessions, platform, yield, read-length stats), plus a link to ENA. An interactive NanoPlot and fastp panel shows read length and quality for quick QC.
(B) Assembly-free overview: A Krona sunburst shows which microbes are present. A paired bar + pie view highlights common antibiotic-resistance, virulence and mobile-element genes for an immediate functional snapshot.
(C) Assembly-based overview: An assembly dashboard summarizes contigs. A table lists recovered genome bins (MAGs) with quality (CheckM2 completeness/contamination) and taxonomy (GTDB-Tk), each linking to a MAG page; nearby plots summarize quality tiers, and a tree view places genomes in context.
(D) Gene-centric results: Widgets consolidate gene-level insights: (1) gene-level Krona after ORF clustering; (2) functional categories from eggNOG/dbCAN; (3) a resistome bar chart (RGI) showing the most prevalent AMR classes.
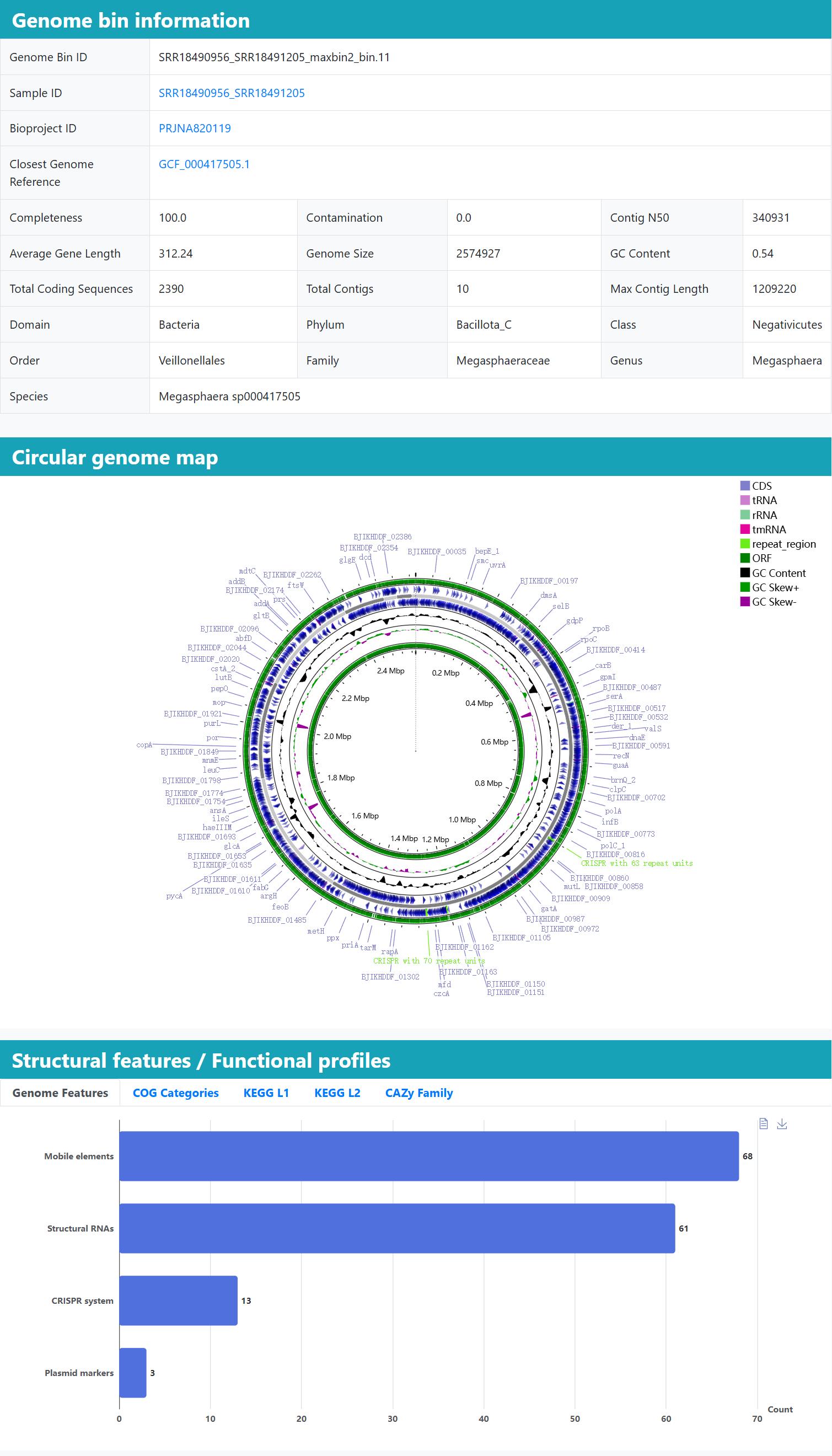
>Detailed information for individual Genome bins
(A) MAG summary table: Bin ID, completeness, contamination, genome size, N50, GC content, contig count and full GTDB taxonomy, with links back to the parent sample and a closest RefSeq genome. (Quality by CheckM2; taxonomy by GTDB-Tk.)
(B) Interactive circular map: A scroll/zoom genome map (CGView) layers coding sequences, rRNAs/tRNAs, repeats, GC-skew and ORF density to help spot genomic islands or rRNA operons.
(C) Genome Features (mobile elements, structural RNAs, CRISPRs); COG Categories, KEGG L1, KEGG L2 for broad-to-fine functions, with Top-N controls and downloads. (Functions from eggNOG; CAZymes via dbCAN; AMR via RGI as applicable.)
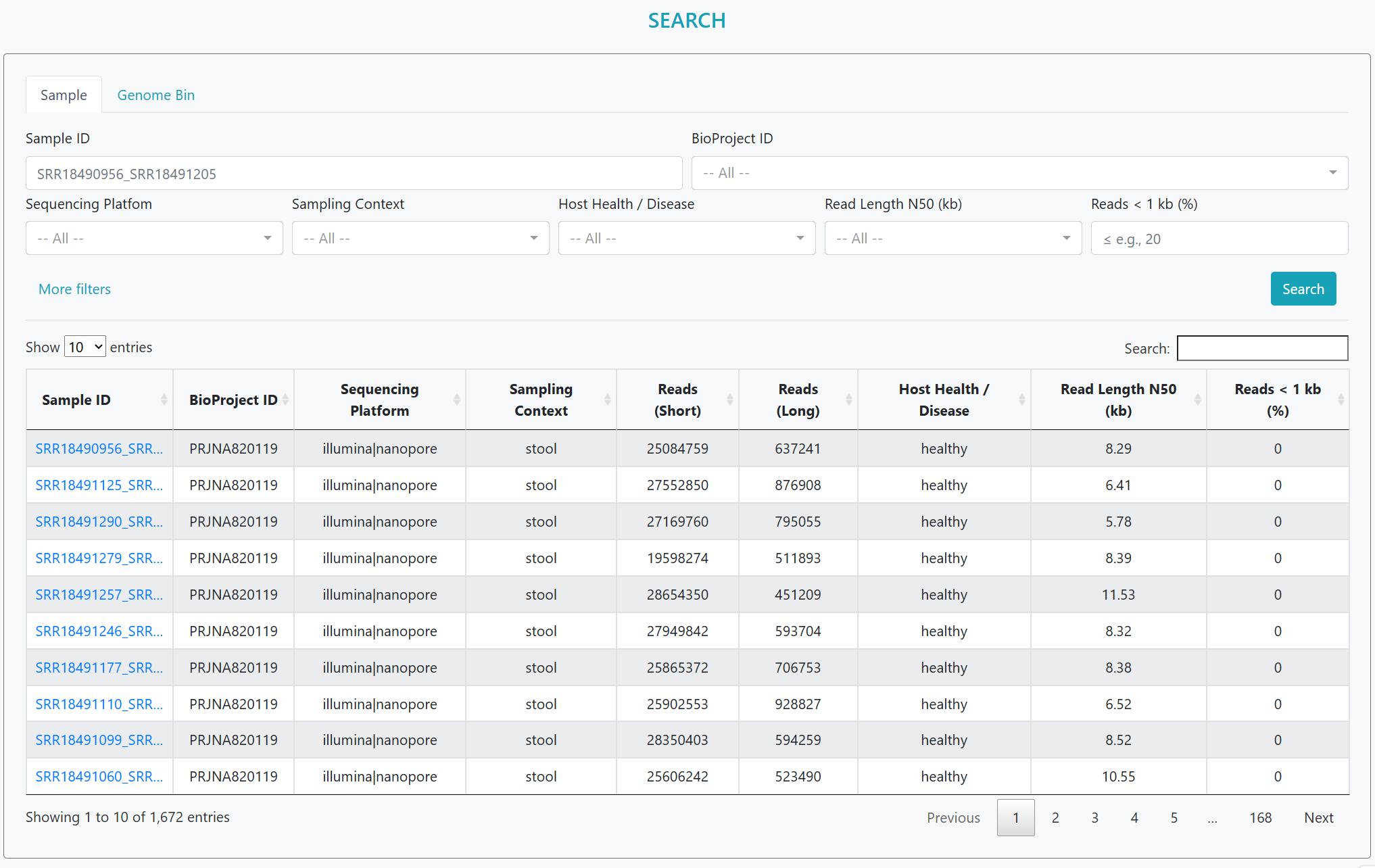
Find what you need without scrolling the whole table. Search across multiple fields and refine with filters.
Step 1 — Start typing. Enter a Sample ID, BioProject ID, or a keyword to seed the query.
Step 2 — Narrow with filters. Use the drop-downs to choose Sequencing platform, Sampling context (body site), Host health/disease, and (optionally) set a Read count range to focus on deeper datasets.
Step 3 — Review & sort. Results update instantly. You can sort any column, use the global search for fuzzy matches, or switch to the Genome bins tab to apply the same filters to MAGs.
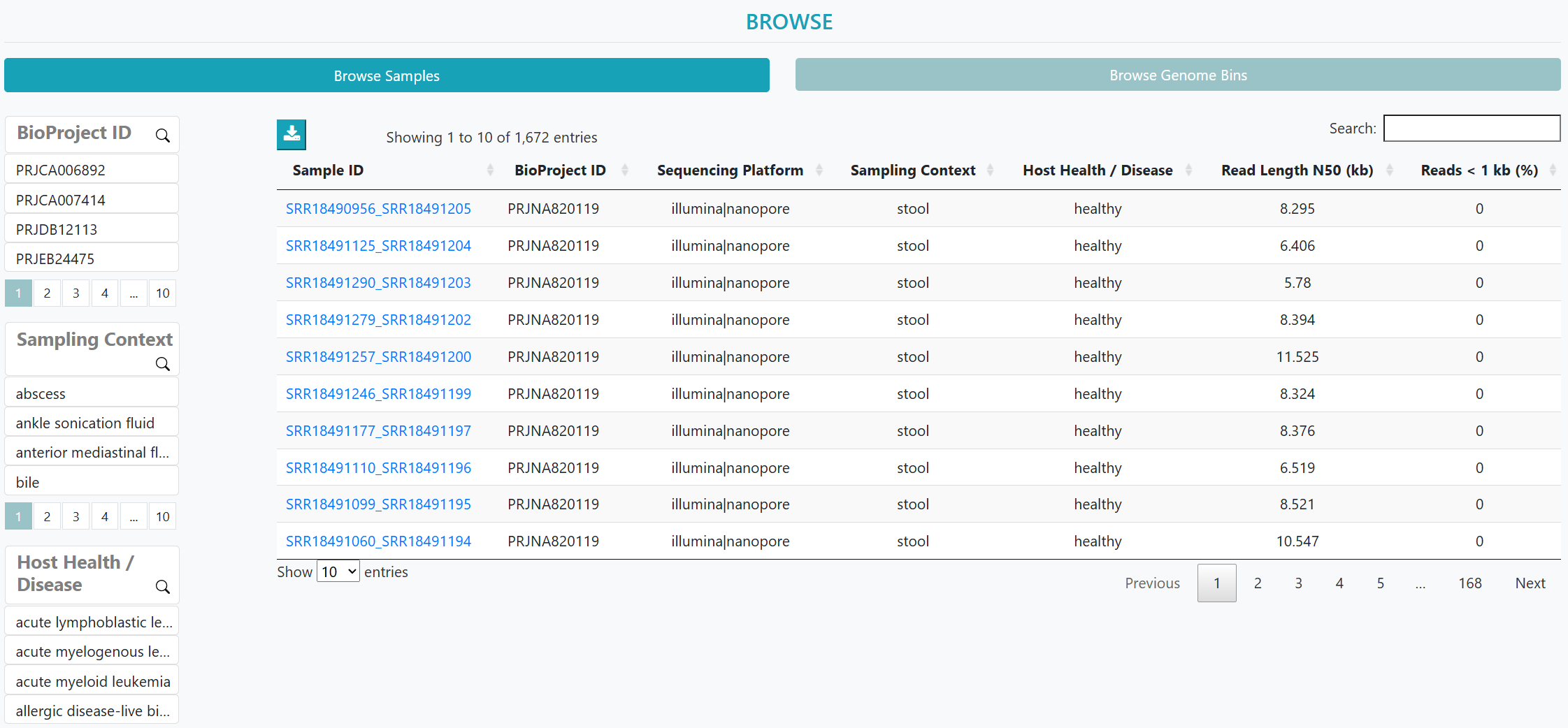
Browse matching samples and genome bins in the interactive tables below. For Samples, filter by BioProject ID, Sampling context, Sequencing platform, and Host health/disease. Select a Sample ID to open details. For Genome bins (MAGs), switch to the Genome bins tab and filter by Taxonomy (GTDB), Completeness/Contamination, and assembly metrics. Select a Bin ID to view the page.
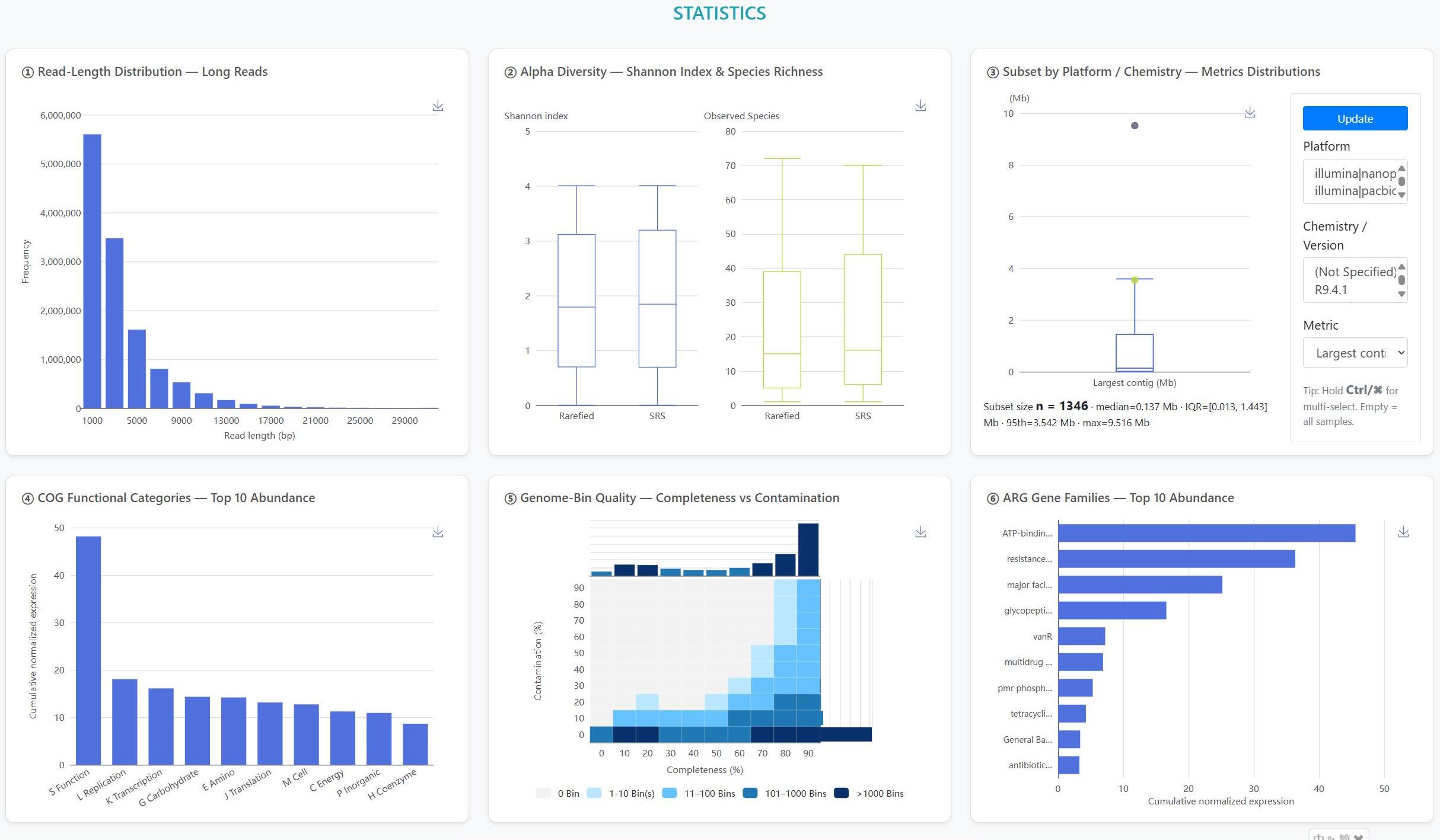
The "Statistics" page gives an at-a-glance view of the collection in six panels: Read length, Diversity, Assembly metrics, COG functions, MAG quality, and AMR overview. Use it to scan trends quickly before diving into details.
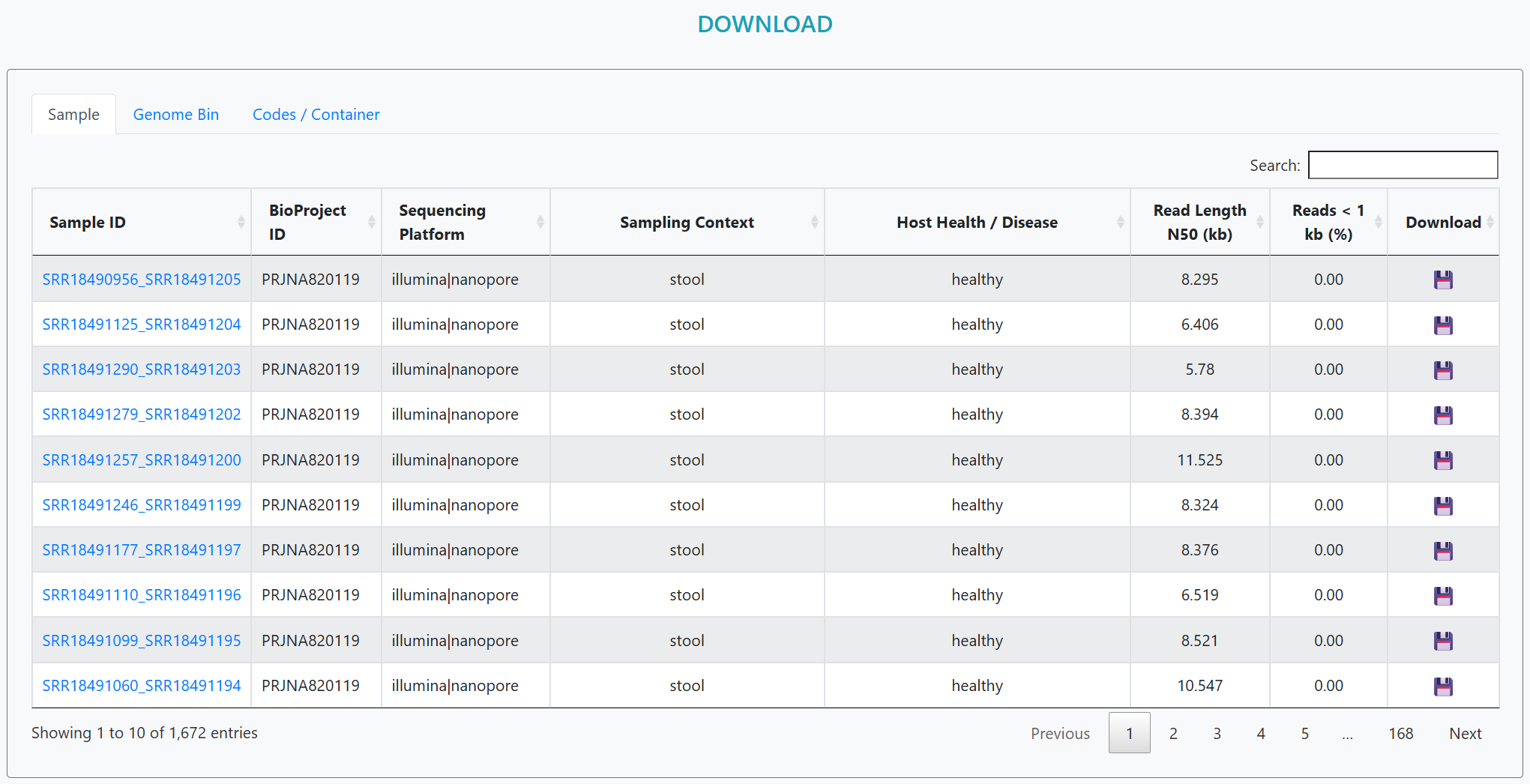
Download assemblies, MAG genomes, and annotation tables in bulk — plus example analysis code. Use the save icons to fetch files for each sample or genome bin (MAG).
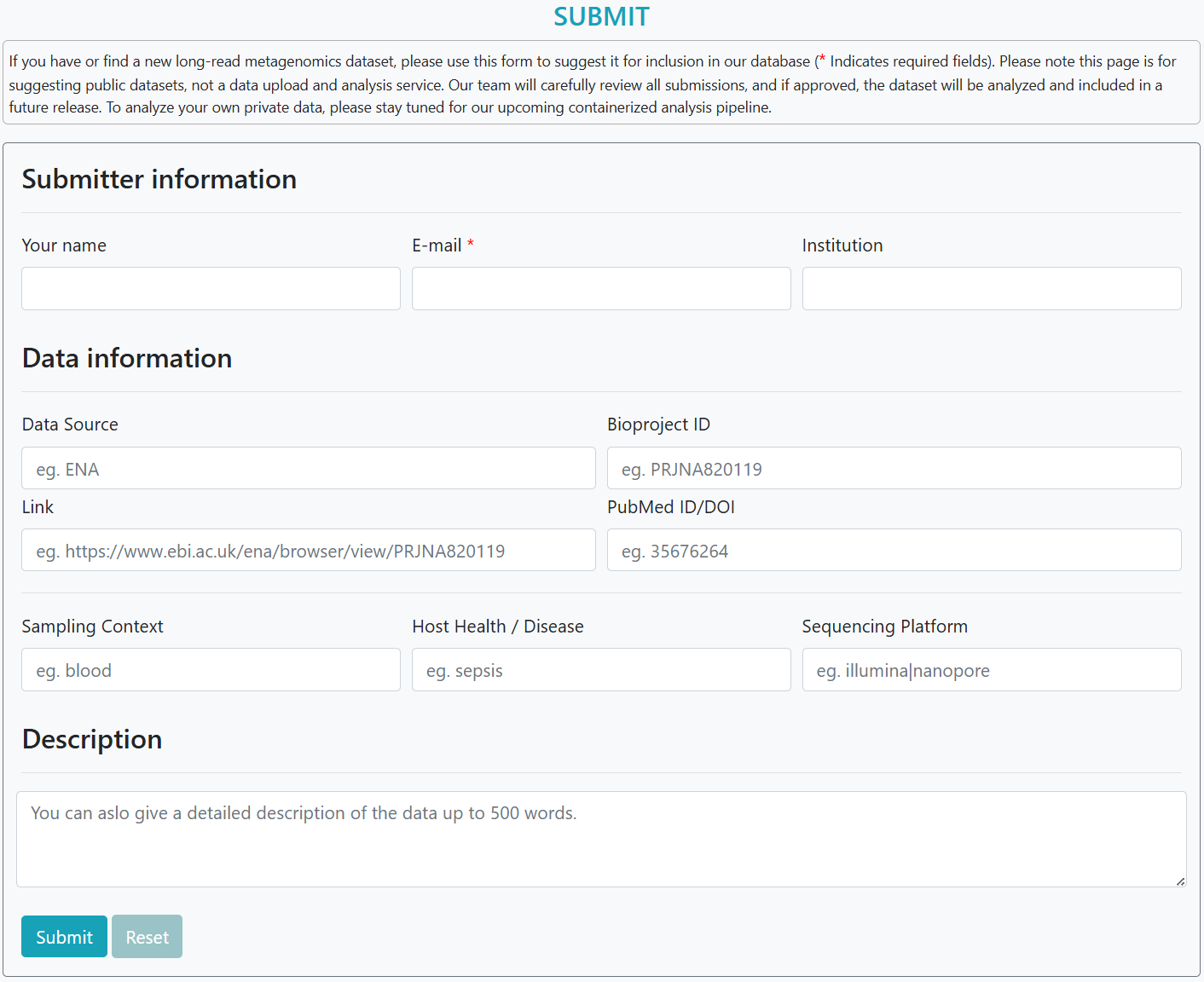
Use "Submit". Provide name, email, institution and study details (BioProject, accession/PubMed/DOI, sampling context, host health, platform; optional ≤500-word note). We verify and run the HLRMDB pipeline; approved data appear in the next release.
See what HLRMDB can do and how to use it—short feature overviews and step-by-step guides.
- Please feel free to contact Prof. Jianbo Pan with respect to any details pertaining to HLRMDB.
-
>Address :
Basic Medicine Research and Innovation Center for Novel Target and Therapeutic Intervention, Ministry of Education, College of Pharmacy, Chongqing Medical University.
No. 1 Yixueyuan Road, Yuzhong District, Chongqing, 400016, P. R. China.
-
>Email :
panjianbo@cqmu.edu.cn